Individual pipeline steps¶
fanc map: Mapping FASTQ files¶
To (iteratively) map FASTQ files directly with FAN-C, use the fanc map command.
Here is a minimal example:
fanc map SRR4271982_chr18_19_1.fastq.gzip bwa-index/hg19_chr18_19.fa \
SRR4271982_chr18_19_1.sam
This command will map the FASTQ file SRR4271982_chr18_19_1.fastq.gzip using the BWA
index specified with -i bwa-index/hg19_chr18_19.fa and output the mapped reads
to SRR4271982_chr18_19_1.sam.
Note
When downloading FASTQ files from SRA using SRAtools, e.g. with fastq-dump, do not
use the -I / --readids option, which appends .1 or .2 to the read name. This
interferes with the sorting and read pairing step in FAN-C. Read names of the two mates
must be identical.
You can change the suffix of the output file to .bam
and fanc map will automatically convert the mapping output to BAM format.
Importantly, fanc map will autodetect if you supply a BWA or Bowtie2 index, so
the following command would use Bowtie2 as a mapper:
fanc map SRR4271982_chr18_19_1.fastq.gzip hg19_chr18_19/hg19_chr18_19 \
SRR4271982_chr18_19_1.sam
You can use additional parameters to control the mapping process:
usage: fanc map [-h] [-m MIN_SIZE] [-s STEP_SIZE] [--trim-front] [-t THREADS]
[-q QUALITY] [-r RESTRICTION_ENZYME] [-k MAX_ALIGNMENTS] [-a]
[-b BATCH_SIZE] [--fanc-parallel] [--split-fastq]
[--memory-map] [--no-iterative] [-tmp]
input [input ...] index output
Positional Arguments¶
- input
File name of the input FASTQ file (or gzipped FASTQ)
- index
Bowtie 2 or BWA genome index base. Index type will be determined automatically.
- output
Output file or folder. When providing multiple input files, this must be the path to an output folder.
Named Arguments¶
- -m, --min-size
Minimum length of read before extension. Default 25.
- -s, --step-size
Number of base pairs to extend at each round of mapping. Default is 10.
- --trim-front
Trim reads from front instead of back.
- -t, --threads
Number of threads used for mapping. Default: 1
- -q, --quality
Mapping quality cutoff. Alignments with a quality score lower than this will be sent to another mapping iteration. Default: 3 (BWA), 30 (Bowtie2)
- -r, --restriction-enzyme
Name (case sensitive) of restriction enzyme used in Hi-C experiment. Will be used to split reads by predicted ligation junction before mapping. You can omit this if you do not want to split your reads by ligation junction. Restriction names can be any supported by Biopython, which obtains data from REBASE (http://rebase.neb.com/rebase/rebase.html). For restriction enzyme cocktails, separate enzyme names with “,”
- -k, --max-alignments
Maximum number of alignments per read to be reported.
- -a, --all-alignments
Report all valid alignments of a read Warning: very slow!.
- -b, --batch-size
Number of reads processed (mapped and merged) in one go per worker. The default 100000 works well for large indexes (e.g. human, mouse). Smaller indexes (e.g. yeast) will finish individual bowtie2 processes very quickly - set this number higher to spawn new processes less frequently.
- --fanc-parallel
Use FAN-C parallelisation, which launches multiple mapper jobs. This may be faster in some cases than relying on the internal paralellisation of the mapper, but has potentially high disk I/O and memory usage.
- --split-fastq
Split FASTQ file into 10M chunks before mapping. Easier on tmp partitions.
- --memory-map
Map Bowtie2 index to memory. Enable if you you system has enough memory to hold the entire Bowtie2 index.
- --no-iterative
Do not use iterative mapping strategy. (much faster, less sensitive).
- -tmp, --work-in-tmp
Copy original file to temporary directory.Reduces network I/O.
Most importantly, assign more threads to the mapping process using the -t parameter.
fanc auto parallelises mapping by spawning multiple mapping processes internally.
This can result in high disk I/O - if you have issues with poor performance,
try using the --mapper-parallel option, which will instead use the multithreading
of your chosen mapping software.
fanc map SRR4271982_chr18_19_1.fastq.gzip bwa-index/hg19_chr18_19.fa \
SRR4271982_chr18_19_1.sam -t 16
By default, fanc auto performs iterative mapping: Reads are initially trimmed to 25bp
(change this with the -m option) before mapping, and then iteratively expanded by 10bp
(change the step size with the -s option) until a unique, high quality mapping location
can be found. The associated quality cutoff is 3 for BWA and 30 for Bowtie2, but can be
changed with the -q parameter.
# expand by 5bp every iteration and be satisfied with lower quality
fanc map SRR4271982_chr18_19_1.fastq.gzip bwa-index/hg19_chr18_19.fa \
SRR4271982_chr18_19_1.sam -t 16 -s 5 -q 10
The iterative mapping process is slower than simple mapping, but can typically
improve mapping efficiency by a few percent, as smaller reads have a lower likelihood of
mismatches due to sequencing errors. Disable iterative mapping with the --no-iterative
parameter.
# do not perform iterative mapping
fanc map SRR4271982_chr18_19_1.fastq.gzip bwa-index/hg19_chr18_19.fa \
SRR4271982_chr18_19_1.sam -t 16 --no-iterative
BWA will automatically split chimeric reads and return both mapping locations. This is
especially useful for Hi-C data, as reads are often sequenced through a ligation junction,
which BWA can often detect automatically. Nonetheless, mapping may be improved by splitting
reads at predicted ligation junctions from the start. To enable this, use the -r parameter
and supply the name of a restriction enzyme (e.g. HindIII or MboI). The name will be used to
look up the enzyme’s restriction pattern, predict the sequence of a ligation junction, and
split reads at the predicted junction before mapping starts. Reads split in this manner will
have an additional attribute in the SAM/BAM file ZL:i:<n> where <n> is an integer denoting
the part of the split read.
# Split reads at HindIII ligation junction before mapping
fanc map SRR4271982_chr18_19_1.fastq.gzip bwa-index/hg19_chr18_19.fa \
SRR4271982_chr18_19_1.sam -t 16 -r HindIII
If you are using Bowtie2, you can additionally use the --memory-map option,
which will load the entire Bowtie2 index into memory to be shared across all Bowtie2 processes. Use
this option if your system has a lot of memory available to speed up the mapping. Finally, if you
are using the -tmp option, which causes fanc auto to perform most pipeline steps in a
temporary directory, you may want to use the --split-fastq option to split the FASTQ files into
smaller chunks before mapping, so you can save space on your tmp partition.
fanc pairs: Generating and filtering read Pairs¶
The fanc pairs command handles the creation and modification of Pairs objects, which represent
the mate pairs in a Hi-C library mapped to restriction fragments. Possible inputs are: two SAM/BAM
files (paired-end reads, sorted by read name), a
HiC-Pro valid pairs file,
a 4D Nucleome pairs file,
or an existing FAN-C Pairs object. For all but the latter, an output file is also required as last
positional argument, for Pairs files the output file is optional. If omitted, filtering is performed
in place.
Here are example commands for the different options.
SAM/BAM files:
fanc pairs output/sam/SRR4271982_chr18_19_1_sort.bam \
output/sam/SRR4271982_chr18_19_2_sort.bam \
output/pairs/SRR4271982_chr18_19.pairs \
-g hg19_chr18_19_re_fragments.bed
4D Nucleome pairs file:
fanc pairs 4d_nucleome.pairs output/pairs/4d_nucleome.pairs \
-g hg19_chr18_19_re_fragments.bed
HiC-Pro valid pairs file:
fanc pairs hic_pro.validPairs output/pairs/hic_pro.pairs \
-g hg19_chr18_19_re_fragments.bed
Existing FAN-C Pairs object:
fanc pairs output/pairs/SRR4271982_chr18_19.pairs
As you can see, the -g parameter is not necessary when proving an existing Pairs object,
as this already has all the fragment information stored in the object. Neither do we need an
output file, as further operations will be performed in place. This primarily applies to
the filtering of read pairs according to various criteria.
Note
Read names of the two mates must be identical. Some read names, especially those obtained
from SRA using SRAtools with the -I / --readids option, have .1 or .2 appended
to the read name. This interferes with the sorting and read pairing step in FAN-C, and you
need to remove those endings before import.
Additional parameters primarily control the filtering of read pairs:
usage: fanc pairs [-h] [-g GENOME] [-r RESTRICTION_ENZYME] [-m] [-u] [-us]
[-q QUALITY] [-c CONTAMINANT] [-i INWARD] [-o OUTWARD]
[--filter-ligation-auto] [-d REDIST] [-l] [-p DUP_THRESH]
[-s STATS] [--reset-filters] [--statistics-plot STATS_PLOT]
[--re-dist-plot RE_DIST_PLOT]
[--ligation-error-plot LIGATION_ERROR_PLOT] [-t THREADS]
[-b BATCH_SIZE] [-S] [-f] [--bwa] [-tmp]
input [input ...]
Positional Arguments¶
- input
IMPORTANT: The last positional argument will be the output file, unless only a single Pairs object is provided. In that case, filtering and correcting will be done in place. Possible inputs are: two SAM/BAM files (paired-end reads, sorted by read name) and an output file; a HiC-Pro pairs file (format: name<tab>chr1<tab>pos1<tab>strand1<tab>chr2<tab>pos2<tab>strand2) and an output file; a pairs file in 4D Nucleome format (https://github.com/4dn-dcic/pairix/blob/master/pairs_format_specification.md) and an output file, or an existing fanc Pairs object. In case of SAM/BAM, HiC-Pro, or 4D Nucleome you must also provide the –genome argument, and if –genome is not a file with restriction fragments (or Hi-C bins), you must also provide the –restriction-enzyme argument.
Named Arguments¶
- -g, --genome
Path to region-based file (BED, GFF, …) containing the non-overlapping regions to be used for Hi-C binning. Typically restriction-enzyme fragments. Alternatively: Path to genome file (FASTA, folder with FASTA, HDF5 file), which will be used in conjunction with the type of restriction enzyme to calculate fragments directly.
- -r, --restriction_enzyme
Name of the restriction enzyme used in the experiment, e.g. HindIII, or MboI. Case-sensitive, only necessary when –genome is provided as FASTA. Restriction names can be any supported by Biopython, which obtains data from REBASE (http://rebase.neb.com/rebase/rebase.html). Separate multiple restriction enzymes with “,”
- -m, --filter-unmappable
Filter read pairs where one or both halves are unmappable. Only applies to SAM/BAM input!
- -u, --filter-multimapping
Filter reads that map multiple times. If the other mapping locations have a lower score than the best one, the best read is kept. Only applies to SAM/BAM input!
- -us, --filter-multimapping-strict
Strictly filter reads that map multiple times. Only applies to SAM/BAM input!
- -q, --filter-quality
Cutoff for the minimum mapping quality of a read. For numbers larger than 1, will filter on MAPQ. If a number between 0 and 1 is provided, will filter on the AS tag instead of mapping quality (only BWA). The quality cutoff is then interpreted as the fraction of bases that have to be matched for any given read. Only applies to SAM/BAM input! Default: no mapping quality filter.
- -c, --filter-contaminant
Filter contaminating reads from other organism. Path to mapped SAM/BAM file. Will filter out reads with the same name. Only applies to SAM/BAM input! Default: no contaminant filter
- -i, --filter-inward
Minimum distance for inward-facing read pairs. Default: no inward ligation error filter
- -o, --filter-outward
Minimum distance for outward-facing read pairs. Default: no outward ligation error filter
- --filter-ligation-auto
Auto-guess settings for inward/outward read pair filters. Overrides –filter-outward and –filter-inward if set. This is highly experimental and known to overshoot in some cases. It is generally recommended to specify cutoffs manually.
- -d, --filter-re-distance
Maximum distance for a read to the nearest restriction site. Default: no RE distance filter
- -l, --filter-self-ligations
Remove read pairs representing self-ligated fragments.Default: no self-ligation filter.
- -p, --filter-pcr-duplicates
If specified, filter read pairs for PCR duplicates. Parameter determines distance between alignment starts below which they are considered starting at same position. Sensible values are between 1 and 5. Default: no PCR duplicates filter
- -s, --statistics
Path for saving filter statistics
- --reset-filters
Remove all filters from the ReadPairs object.
- --statistics-plot
Path for saving filter statistics plot (PDF)
- --re-dist-plot
Plot the distribution of restriction site distances of all read pairs (sum left and right read).
- --ligation-error-plot
Plot the relative orientation of read pairs mapped to the reference genome as a fraction of reads oriented in the same direction. Allows the identification of ligation errors as a function of genomic distance.
- -t, --threads
Number of threads to use for extracting fragment information. Default: 1
- -b, --batch-size
Batch size for read pairs to be submitted to individual processes. Default: 1000000
- -S, --no-check-sorted
Assume SAM files are sorted and do not check if that is actually the case
- -f, --force-overwrite
If the specified output file exists, it will be overwritten without warning.
- --bwa
Use filters appropriate for BWA and not Bowtie2. This will typically be identified automatically from the SAM/BAM header. Set this flag if you are having problems during filtering (typically 0 reads pass the filtering threshold).
- -tmp, --work-in-tmp
Work in temporary directory
Filtering¶
fanc pairs provides a lot of parameters for filtering read pairs according to different
criteria. By default, if not specified otherwise, no filtering is performed on the read pairs
(passthrough). Typically, however, you will at least want to filter out unmappable (-m)
and multimapping reads (-u or -us). It is also a good idea to filter by alignment
quality (-q <n>). Good cutoffs for Bowtie2 and BWA are 30 and 3, respectively. If you suspect
your Hi-C library to be contaminated by DNA from a different organism, you can align your
original reads to a different genome and pass the resulting SAM/BAM file to the -c
parameter (ensure no unmappable reads are in the file!).
This will filter out all reads that have a valid alignment in the putative
contaminants genome (by qname). All of the above filters operate on single reads, but will
filter out the pair if either of the reads is found to be invalid due to a filtering criterion.
Warning
The -u, -us, -q, and -c filter MUST be applied when loading read pairs
from SAM/BAM file, and cannot be added later!
fanc pairs output/sam/SRR4271982_chr18_19_1_sort.bam \
output/sam/SRR4271982_chr18_19_2_sort.bam \
output/pairs/SRR4271982_chr18_19.pairs \
-g hg19_chr18_19_re_fragments.bed \
-us \
-q 3
An additional set of filters operates on the properties of the read pair. You may want to
filter out self-ligated fragments, which provide no spatial information with the -l
parameter. As Hi-C experiments generally rely on PCR amplification, it is expected to find
a lot of PCR duplicates in the library. You can filter those with the -p <n> parameter,
where <n> denotes the distance between the start of two alignments that would still be
considered a duplicate. Normally you would use 1 or 2, but you can use higher values to be
more strict with filtering.
Example:
fanc pairs output/pairs/SRR4271982_chr18_19.pairs \
-l # filter self-ligated fragments \
-p 2 # filter PCR duplicates mapping within 2bp
Depending on the experimental setup, it is sometimes expected to find valid Hi-C alignments
near restriction sites. You can filter read pairs for their (cumulative) distance to the
nearest restriction sites using the -d parameter. To determine that cutoff, or to detect
any issues with the Hi-C library, you can first use the --re-dist-plot parameter. Note
that this will only plot a sample of 10,000 read pairs for a quick assessment:
fanc pairs --re-dist-plot re-dist.png output/pairs/SRR4271982_chr18_19.pairs
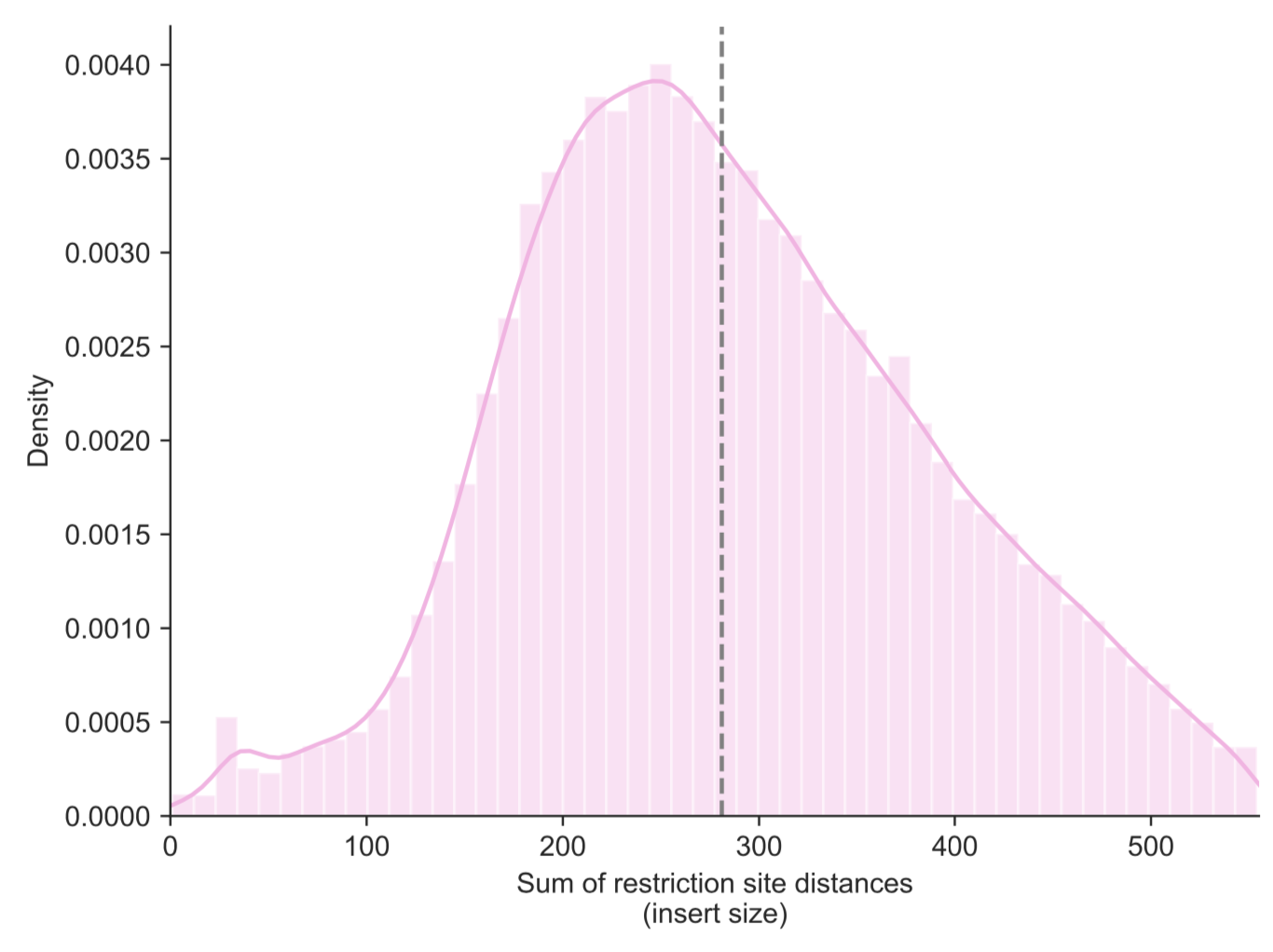
Jin et al. (2013) have identified
several errors that stem from incomplete digestion and which can be identified from different
types of ligation products. You can filter these using the -i <n> and -o <n> parameters,
for the inward and outward ligation errors, respectively. If you need help finding a good
cutoff, you may use the --ligation-error-plot parameter.
fanc pairs --ligation-error-plot ligation-err.png output/pairs/SRR4271982_chr18_19.pairs
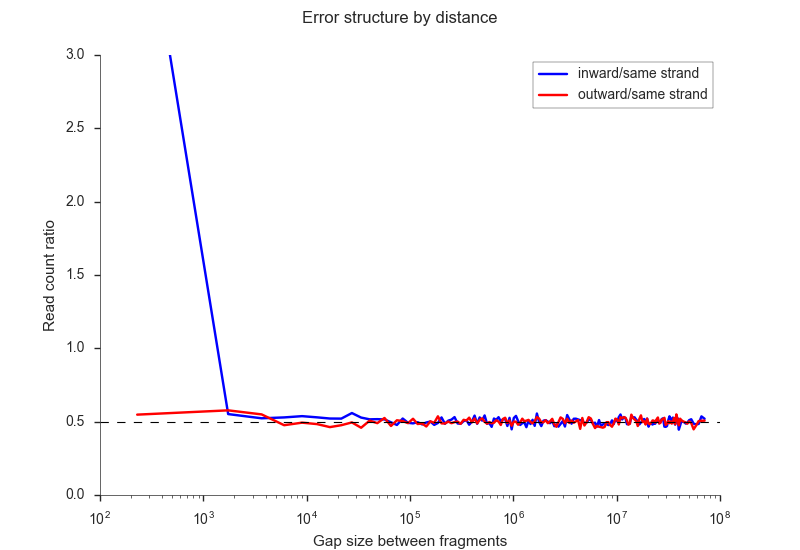
Otherwise 1-10kb are often a reasonable cutoffs. You can also let FAN-C attempt to find suitable
cutoffs based on the over-representation of certain ligation products using the
--filter-ligation-auto parameter, but this is not always 100% reliable.
Finally, you can output the filtering statistics to a file or plot using the -s and
--statistics-plot parameters, respectively.
fanc pairs output/pairs/SRR4271982_chr18_19.pairs \
--statistics-plot pairs.stats.png
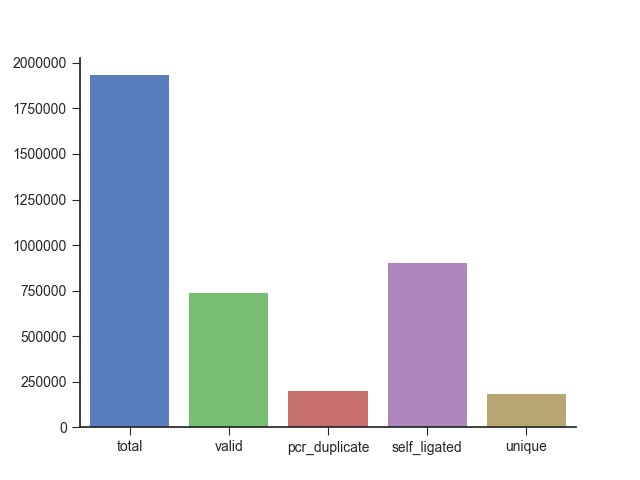
fanc hic: Generating, binning, and filtering Hic objects¶
The fanc hic command is used to generate fragment-level and binned Hi-C matrices.
You can use FAN-C Pairs files as input for fanc hic:
fanc hic output/pairs/SRR4271982_chr18_19.pairs output/hic/fragment_level.hic
Without additional parameters, this will generate a fragment-level Hic object and exit. Multiple Pairs files will be converted into fragment-level Hic objects which are then merged into a single object.
If you already have a fragment-level Hic file and you want to bin it, or perform filtering or matrix balancing, you can also use this as input:
fanc hic output/hic/fragment_level.hic output/hic/binned/example_1mb.hic -b 1mb
You have to explicitly provide the binning, filtering and correcting parameters, otherwise the command will exit after it has obtained a single fragment-level Hic object. Here is an overview of all parameters:
usage: fanc hic [-h] [-b BIN_SIZE] [-l FILTER_LOW_COVERAGE]
[-r FILTER_LOW_COVERAGE_RELATIVE] [-a] [-d FILTER_DIAGONAL]
[--marginals-plot MARGINALS_PLOT] [--reset-filters]
[--downsample DOWNSAMPLE] [--subset SUBSET] [-i] [-k] [-n]
[-m NORM_METHOD] [-w] [-c] [--only-inter] [-s STATS]
[--statistics-plot STATS_PLOT]
[--chromosomes CHROMOSOMES [CHROMOSOMES ...]] [-f]
[-t THREADS] [--deepcopy] [-tmp]
input [input ...]
Positional Arguments¶
- input
IMPORTANT: The last positional argument will be the output file, unless only a single Hic object is provided. In that case, binning, filtering and correcting will be done in place. Input files. If these are FAN-C Pairs objects (see “fanc pairs”), they will be turned into Hic objects. Hic objects (also the ones converted from Pairs) will first be merged and the merged object will be binned, filtered and corrected as specified in the remaining parameters.
Named Arguments¶
- -b, --bin-size
Bin size in base pairs. You can use human-readable formats,such as 10k, or 1mb. If omitted, the command will end after the merging step.
- -l, --filter-low-coverage
Filter bins with low coverage (lower than specified absolute number of contacts)
- -r, --filter-low-coverage-relative
Filter bins using a relative low coverage threshold (lower than the specified fraction of the median contact count)
- -a, --low-coverage-auto
Filter bins with “low coverage” (under 10% of median coverage for all non-zero bins)
- -d, --diagonal
Filter bins along the diagonal up to this specified distance. Use 0 for only filtering the diagonal.
- --marginals-plot
Plot Hi-C marginals to determine low coverage thresholds.
- --reset-filters
Remove all filters from the Hic object.
- --downsample
Downsample a binned Hi-C object before filtering and correcting. Sample size or reference Hi-C object. If sample size is < 1,will be interpreted as a fraction of valid pairs.
- --subset
Comma-separated list of regions that will be used in the output Hic object. All contacts between these regions will be in the output object. For example, “chr1,chr3” will result in a Hic object with all regions in chromosomes 1 and 3, plus all contacts within chromosome 1, all contacts within chromosome 3, and all contacts between chromosome 1 and 3. “chr1” will only contain regions and contactswithin chromosome 1.
- -i, --ice-correct
DEPRECATED. Use ICE iterative correction on the binned Hic matrix
- -k, --kr-correct
DEPRECATED. Use Knight-Ruiz matrix balancing to correct the binned Hic matrix
- -n, --normalise
Normalise Hi-C matrix according to –norm-method
- -m, --norm-method
Normalisation method used for -n. Options are: KR (default) = Knight-Ruiz matrix balancing (Fast, accurate, but memory-intensive normalisation); ICE = ICE matrix balancing (less accurate, but more memory-efficient); VC = vanilla coverage (a single round of ICE balancing);VC-SQRT = vanilla coverage square root (reduces overcorrection compared to VC)
- -w, --whole-matrix
Correct the whole matrix at once, rather than individual chromosomes.
- -c, --restore-coverage
Restore coverage to the original total number of reads. Otherwise matrix entries will be contact probabilities.
- --only-inter
Calculate bias vector only on inter-chromosomal contacts. Ignores all intra-chromosomal contacts. Always uses whole-matrix balancing, i.e. implicitly sets -w
- -s, --statistics
Path for saving filter statistics
- --statistics-plot
Path for saving filter statistics plot (PDF)
- --chromosomes
Limit output Hic object to these chromosomes. Only available in conjunction with “-b” option.
- -f, --force-overwrite
If the specified output file exists, it will be overwritten without warning.
- -t, --threads
Number of threads (currently used for binning only)
- --deepcopy
Deep copy Hi-C file. Can be used
- -tmp, --work-in-tmp
Work in temporary directory
Binning¶
You can use the -b parameter to bin the fragment-level Hi-C matrix. You can either use
integers (1000000) or common abbreviations (1Mb). The filtering steps outlined below only
apply to binned Hic matrices.
Filtering and balancing¶
fanc hic provides a few filtering options. Most likely you want to apply a coverage filter
using -l to specify a coverage threshold in absolute number of pairs per bin, or -r to
apply a coverage threshold based on a fraction of the median number of pairs pair bin. -a is
simply a preset for -r 0.1.
For some applications it might be useful to remove the prominent Hi-C diagonal. You can use the
-d <n> parameter to remove all pairs in pixels up to a distance of n from the diagonal.
You can balance your Hi-C matrices using the -n parameter. By default, this applies Knight-Ruiz
(KR) balancing to each chromosome. You can opt for another normalisation method using
--norm-method, for example --norm-method ice for ICE balancing, or --norm-method vc
for vanilla coverage normalisation (which in this case is equivalent of a single ICE iteration).
We typically recommend KR balancing for performance reasons. Each chromosome in the matrix is
corrected independently, unless you specify the -w option.
Note
The default normalisation mode is per chromosome, and the FAN-C analysis functions assume
this has been used. In the majority of cases, when you are working with intra-chromosomal
data, this is what you want. Some commands have a --whole-genome option (or equivalent)
to apply the analysis to the whole matrix instead of individual chromosomes. Only use this
option if you have corrected the whole matrix at once (-w), and if you understand the
effect of whole-matrix analysis on your results, compared to per-chromosome analysis!
When you are working with inter-chromosomal data, the chromosome’s bias vectors are re-used
for normalisation. This is likely not the most suitable normalisation for your inter-chromosomal
data! If you are interested in inter-chromosomal data specifically, you may want to use
whole-genome correction, and possibly also exclude the intra-chromosomal contacts. You can do
that using the --only-inter option.
By default the corrected matrix entries correspond to contact
probabilities. You can use the --restore-coverage option to force matrix entries in a
chromosome to sum up to the total number of reads before correction.
Finally, you can output the filtering statistics to a file or plot using the -s and
--statistics-plot parameters, respectively.